22 / 06 / 12
RNA-seq转录组数据的比对
RNA-seq常见比对软件的使用,包括TopHat2、Hisat2和STAR。
目标:
- 掌握高通量的比对算法。
- 能够下载参考基因组序列、基因注释文件建立index。
- 完成序列比对及BAM文件的转换索引,理解含义。
- 理解不同比对软件的优劣并能合理选择。

本节测试数据下载方式: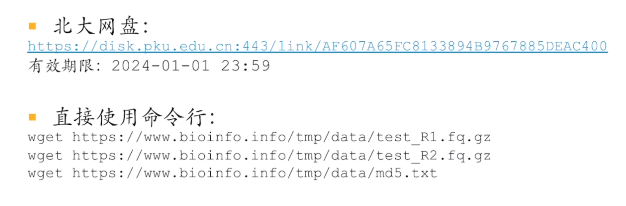
Alignment和Mapping的区别?
RNA-seq数据分析的相关软件:
质控:fastqc ,multiqc , trimmomatic, cutadapt, trim-galore
比对:star , hisat2 , tophat , bowtie2 , bwa , subread
定量:htseq , bedtools, salmon, Kallisto
1、拟南芥参考基因组/转录组的下载
下载网址:http://ftp.ensemblgenomes.org/pub/plants/release-53/fasta/arabidopsis_thaliana/dna/
一般我们下载全染色体的基因组文件:http://ftp.ensemblgenomes.org/pub/plants/release-53/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
拟南芥基因组索引文件:http://ftp.ensemblgenomes.org/pub/plants/release-53/fasta/arabidopsis_thaliana/dna_index/
一般我们下载*toplevel.fa.gz文件,为参考基因组完整文件,其他rm,sm,和分开染色体得文件;sm和rm的意义可看README文件。
教程:https://blog.csdn.net/u010608296/article/details/121438333

Ensembl和NCBI基因组下载,基因序列下载查看.pdf
拟南芥基因组注释gff3文件下载:http://ftp.ensemblgenomes.org/pub/plants/release-53/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.53.gff3.gz
拟南芥基因组注释gtf文件下载:http://ftp.ensemblgenomes.org/pub/plants/release-53/gtf/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.53.gtf.gz
2、TopHat和TopHat2
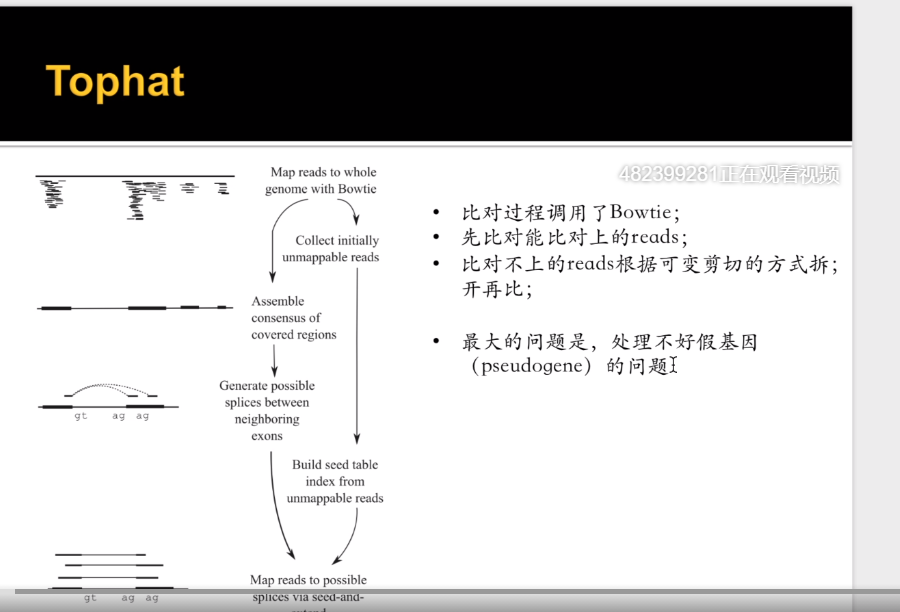
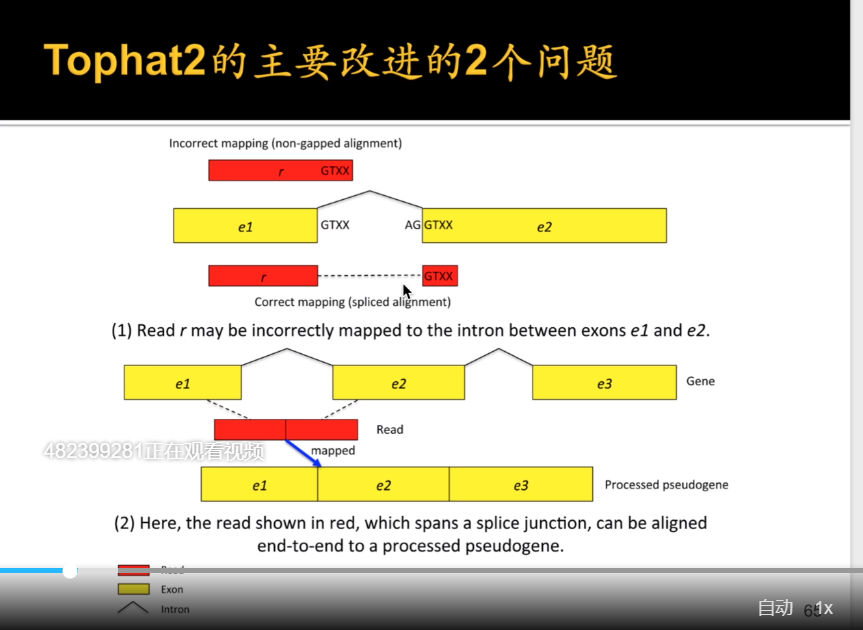
tophat2需要python2的环境。
首先需要用bowtie2构建基因组index:
bowtie2-build --thread 6 ref_hg38.fa ref_hg38.fa > bt2_index.log 2>&1 & # 主要设置一个thread参数即可,这是线程参数。第一个ref_hg38.fa表示那个基因组,第二个表示的是名字。 less -S bt2_index.log # 查看log文件。

log的最后一行会提示用了多长时间。
tophat2 -G ./xx.gtf -o ./temp_tophat2 ./index.xx.fa ../test_R1_cutadapt.fa.gz .. /test_R2_cutadapt.fa.gz & # -G参数对应的是基因组注释文件。-o对应的是tophat2输出结果。 # tophat2也可以加一个-p参数。比如tophat2 -p 6,6个核。
tophat2的输出结果:
可以看到那里是deletions,那里是insertions。
比较好的是,直接给出了bam文件。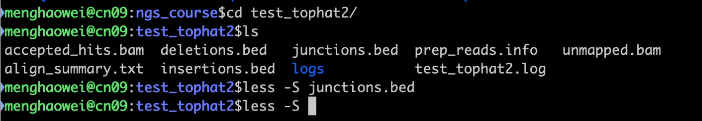
# 查看文件头 samtools view -H accepted_hits.bam # -H这个参数的作用是输出文件头。 # 查看比对结果 samtools view accepted_hits.bam | less -S
bam文件文件头:
SO: coordinate表示这个bam文件已经排过序了。而且是按照基因组坐标进行排序的。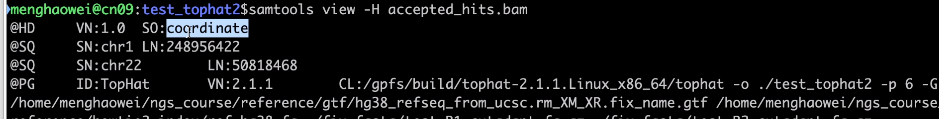
2、STAR:Spliced Transcripts Alignment to a Reference
STAR的优势在于快,能够快速的mapping。
缺点在于占用内存巨大,human的mapping需要28G~32 GB左右的RAM。
STAR的出现,完全是为了ENCODE计划。(ENCODE: Encyclopedia of DNA Elements)
运行一个程序是32 GB内存,运行100个程序也是32 GB内存,因此大型项目都喜欢用STAR软件。
STAR软件的策略:
STAR首先将一个reads切成两份,找到全基因组上对应的位置。
通过打分算法,把邻近的全基因组符合的seeds都拼接在一起,形成mappting结果。
优势在于支持并行计算。
STAR的比对质量有时候还不如TopHat2。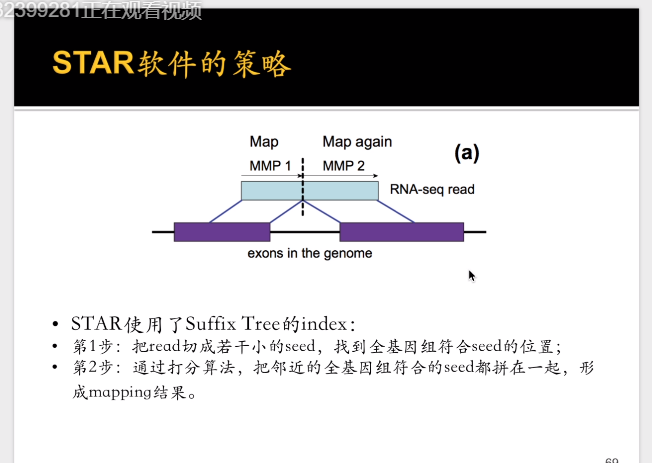
mkdier temp_star_index #首先建立star index STAR --runThreadN 12 --runMode genomeGenerate \ # runMode总共有两种。一种是genomeGenerate,一种是mapping。 --genomeDir /home/menghaowei/ngs_course/reference/STAR_index \ # 需要创建的index位置。 --genomeFastaFiles /home/menghaowei/ngs_course/reference/STAR_index/ref_ref_hg38.fa \ 参考基因组。 --sjdbGTFfile /home/menghaowei/ngs_course/reference/gtf/hg38xxxx.gtf \ # 基因组注释文件。 --sjdbOverhang 150 & # 分析的是100 bp的reads,就写100。分析的是150 bp的reads就写150。目前illumina的都是150 bp的reads。
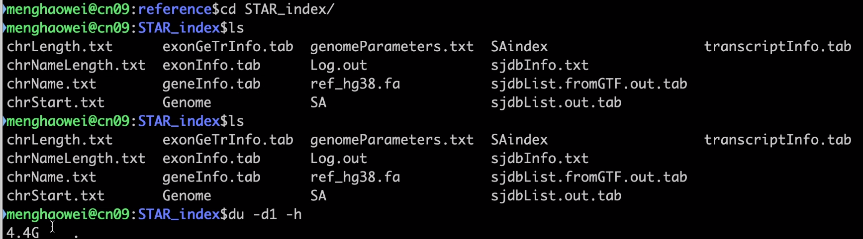
查看STAR index:
只用了人的两条染色体,建立出来的index大小居然有4.4 G。恐怖。
du -dl -h 查看大小
less log.out tail log.out #查看日志尾部。
STAR进行比对:
# 这是常用的参数。一般不要轻易的更改。 STAR \ --genomeDir /home/menghaowei/ngs_course/reference/STAR_index \ --runThreadN 6 \ --readFileIn ./fix.fastq/test_R1_cutadapt.fq.gz ./fix.fastq/test_R2_cutadapt.fq.gz \ --readFilescommand zcat \ --outFileNamePrefix ./bam/test-STAR \ --outSAMtype BAM Unsorted \ # 输出的文件类型。不排序的BAM。 --outSAMstrandField intronMotif \ --outSAMattributes All \ # 这个地方一定要选All。 --outFilterIntronMotifs RemoveNoncanonical > ./bam/test_STAR.log 2>&1 &
查看STAR结果:
ls less -S test_STAR.log
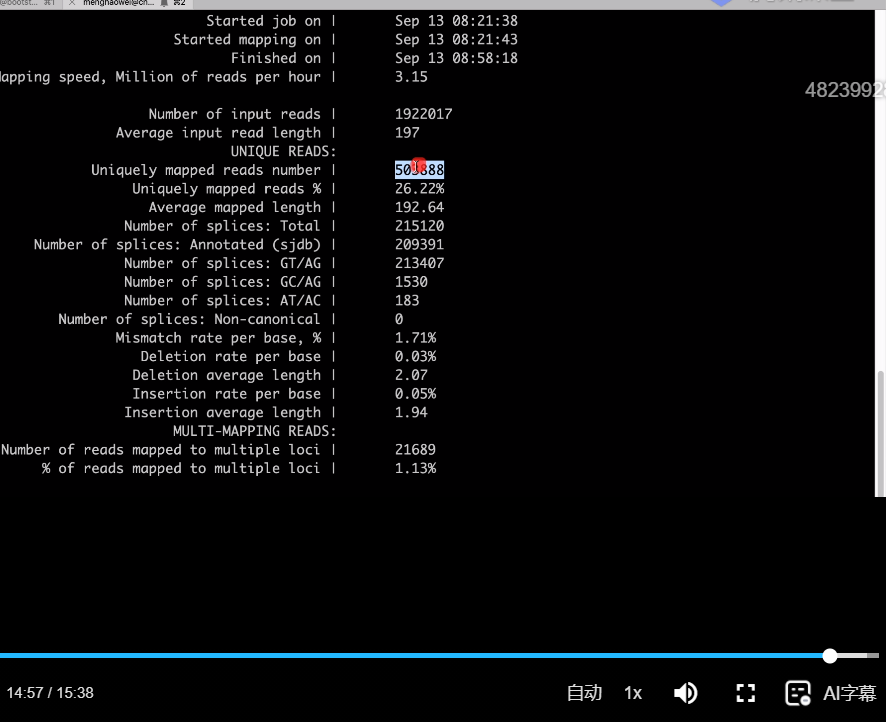
最后,STAR一般适合大型项目。准确率还可以。
3、HISAT和HISAT2
HISAT2可以同时比对DNA和RNA。
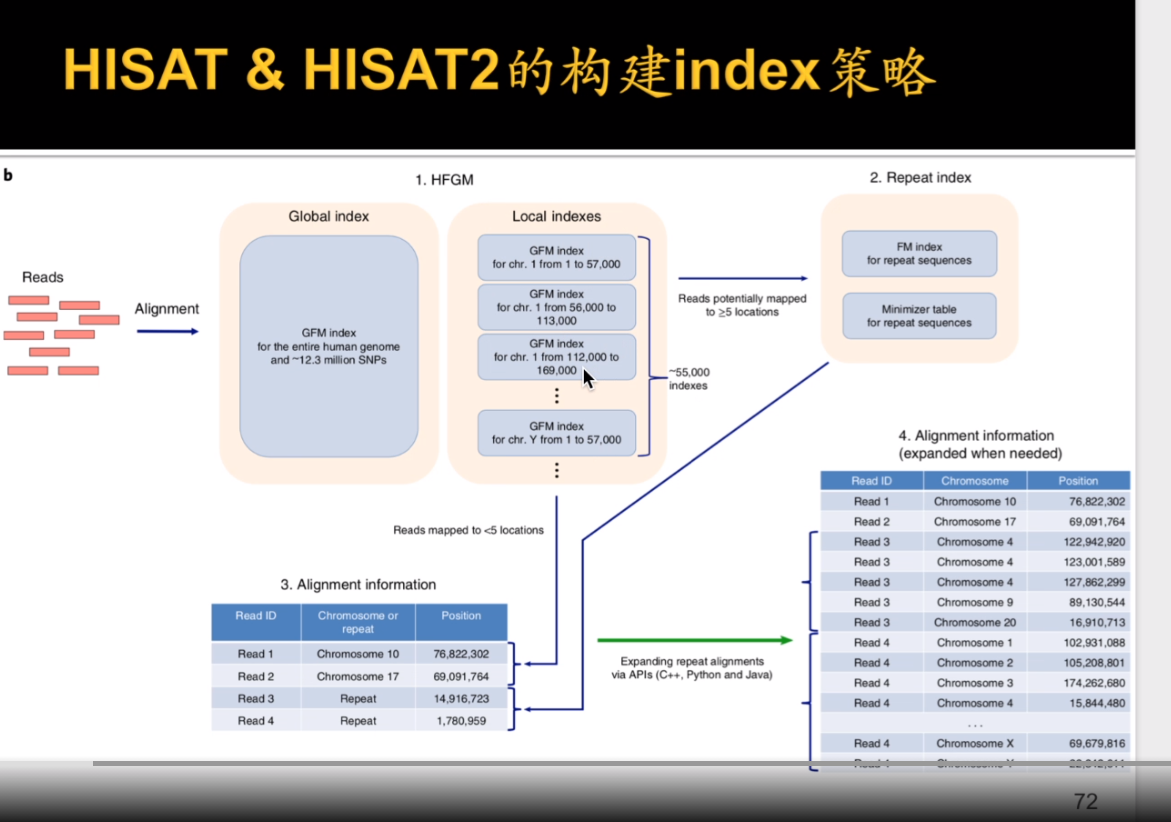
比对软件的时间线:
hisat2-build -t 6 ref_hg38.fa ref_hg38.fa >hisat2_build.log 2>&1 & # 和bowtie2-build很像。 # hisat2比对 hisat2 -p 12 \ # 12个核心。 -x /home/menghaowei/ngs_course/reference/hisat2_index/ref_hg38.fa \ -1 ./fix.fastq/test_R1_cutadapt.fq.gz \ -2 ./fix.fastq/test_R2_cutadapt.fa.gz \ -S ./bam/test_hisat2.sam > ./bam/test_hisat2.log 2>&1 & # -S是输出的SAM文件。
如果有SNP,需要给出SNP信息: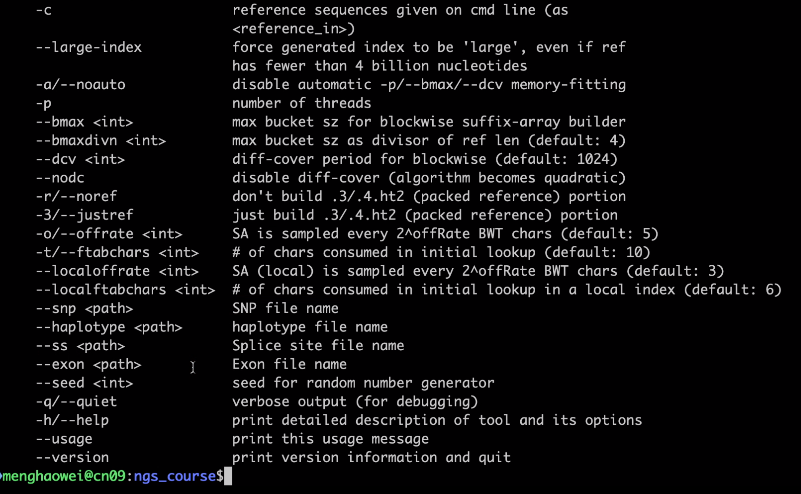
HISATA2 index的构建:
可以增加SNP信息和可变剪切信息。

SNP可在UCSC网站下载。
hisat2_extract_exons.py hg38_refseq.gtf > hg38_refseq.exon & # 从GTF注释文件中提取外显子信息。这个脚本在hisat2中已经包含了。 hisat2_extract_splice_sites.py hg38_refseq.gtf > hg38_refseq.ss & #提取剪切信息。 hisat2_extract_snps_haplotypes_UCSC.py ref_hg38.fa snp151Common.txt snp151Common & #第二个snp151Common是生成的snp文件名称。 hisat2_build -p 6 --snp snp151Common.snp --haplotype snp151Common.haplotype --exon hg38_refseq.exon --ss hg38_refseq.ss ref_hg38.fa.snp_gtf > hisat2_build.log 2>&1 &
SAM文件是不可以的,需要转换为按基因组排序的BAM文件。
samtools sort -O BAM -o ngs_test.sort.bam -@6 -m 2G -T ngs_test.sort.bam.temp test_hisat2.sam & # -O生成的格式为BAM,6个线程,每个线程2G运行内存。生成的文件名称ngs_test.sort.bam.temp。 # 查看生成的BAM文件排序结果 samtools view -h ngs_test.sort.bam | less -S
BAM排序结果: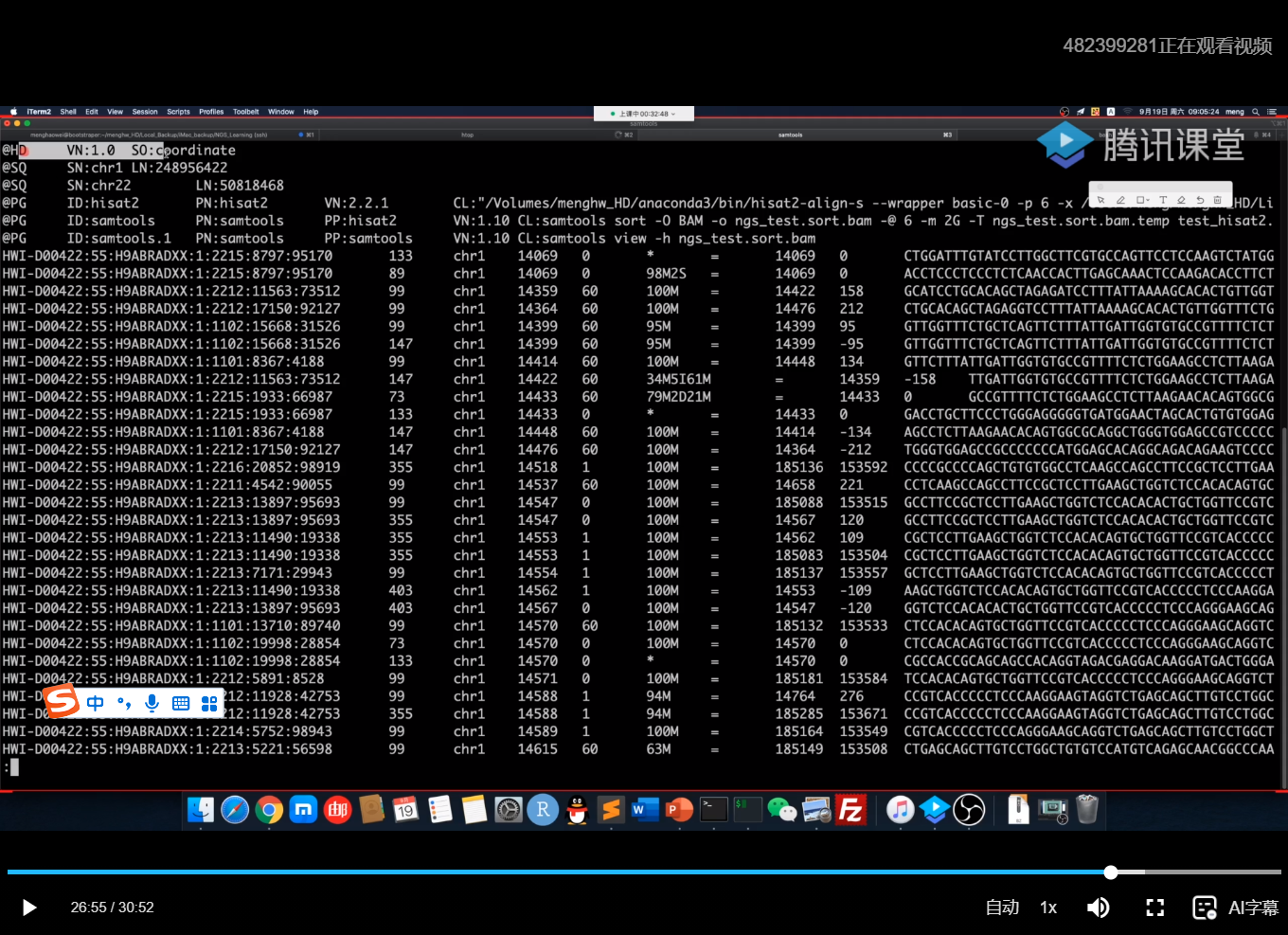
第一行可以看到SO: coordinate,表示已经排过序了。