22 / 06 / 12
使用FastQC、cutadapt进行数据质控
使用FastQC、cutadapt进行数据质控。包括相关的软件安装和使用。
1、NASA示例代码-FastQC【推荐首先阅读】
https://mp.weixin.qq.com/s/2Xo-a3p47kg2feawgeynOg https://github.com/nasa/GeneLab_Data_Processing/tree/master/RNAseq/GLDS_Processing_Scripts
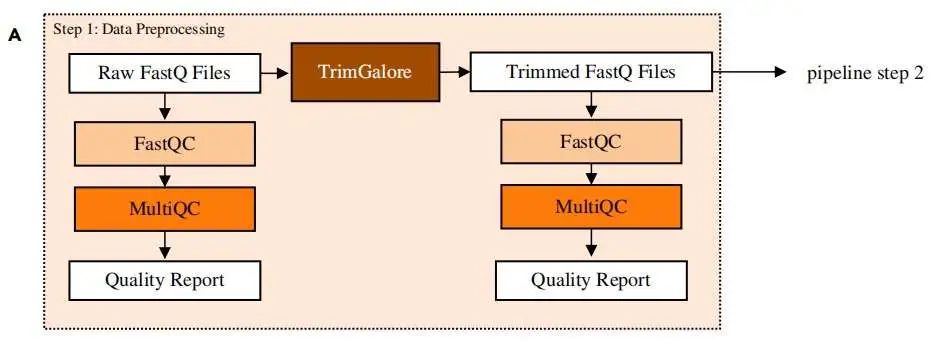
fastqc 代码及结果文件:
fastqc -o /path/to/output/directory \ -t number_of_threads \ /path/to/input/files
multiqc 整合质控结果:
multiqc -o /path/to/output/directory \ /path/to/fastqc/output/files
2、【孟浩巍课程笔记】转录组数据的质控
fastqc安装:
# 首先需要安装conda。 # 不推荐直接安装Anaconda,太过于臃肿。安装miniconda就好了。 conda install fastqc #需要先安装conda。安装conda后非常方便。但是conda比较慢,于是有人开放了mamba。 conda install mamba mamba install fastqc # mamba安装会快很多,能用mamba就用mamba。目前mamba不太好用,可能因为一些更新问题(2022.06.12)。 conda create -n RNA-seq python=3.7 #创建一个专门的环境。 mamba create -n RNA-seq python=3.7 #使用mamba创建一个RNA-seq的专门环境。 conda activate RNA-seq #激活环境。 conda deactivate #退出当前环境。
conda、miniconda和Anaconda的区别:Miniconda 只安装了 Conda 核心,而 Anaconda 除此之外还安装了完整的 Anaconda 的发行包,换句话说后者初始安装的包更多,因此安装文件体积也更大。
https://zhuanlan.zhihu.com/p/28610806 https://blog.csdn.net/qq_18668137/article/details/80807829
一定要先要先创建单独的环境,然后激活环境,再安装软件。将生信软件都安装在这个环境中好了。
查看fastq格式的测序文件:
zcat test_R1.fq.gz | less -S #zcat解压,通过管道服务放到一行(- S参数)。 # zcat命令:Zcat是一个命令行实用程序,用于查看压缩文件的内容,而无需对其进行解压缩。
fastq格式: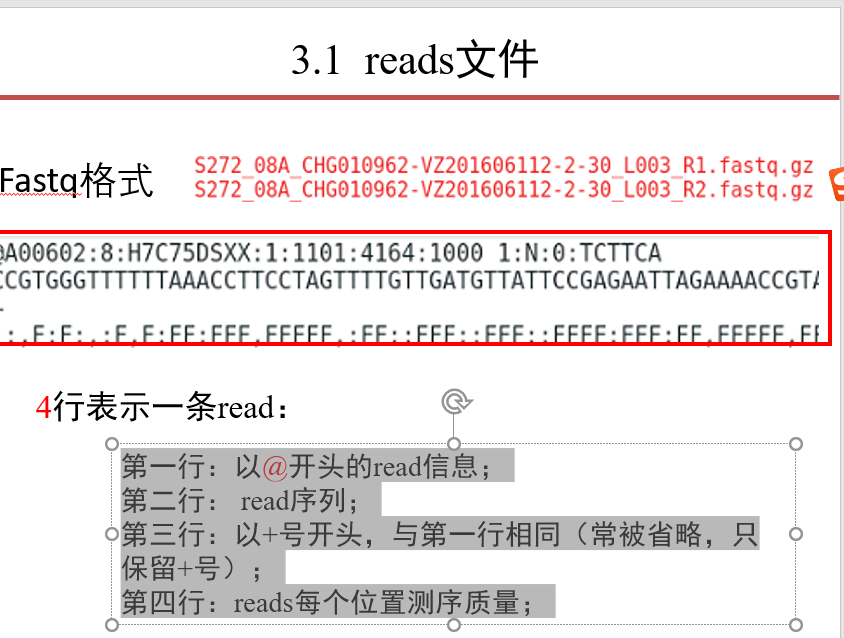
- 第一行:以@开头的read信息;
- 第二行: read序列;
- 第三行:以+号开头,与第一行相同(常被省略,只保留+号);
- 第四行:reads每个位置测序质量;
fastqc语法
fastqc -t 20 -o test_out/ ./raw.fastq/test_R*.gz & # -t表示20个线程,-o表示的是fastqc输出文件夹。./raw.fastq/test_R*.gz指的是输入文件。最后加一个&,表示在后台运行。之后点击回车键就可以开始分析了。 top #该命令可以查看程序的后台运行情况。 kill 123 #终止运行某一个后台程序。
SRA数据库及Linux本地下载
SRA Toolkit安装:
参考连接:https://github.com/ncbi/sra-tools/wiki/02.-Installing-SRA-Toolkit
For Ubuntu,
wget --output-document sratoolkit.tar.gz http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
For CentOS,
wget --output-document sratoolkit.tar.gz http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz
解压文件:
tar -vxzf sratoolkit.tar.gz
添加环境变量:
echo "export PATH=\$PATH:PWD/sratoolkit.2.9.2-ubuntu64/bin" >> ~/.bashrc # echo命令是在屏幕中输出一段字符,该代码的啥意思是,将export PATH=\$PATH:PWD/sratoolkit.2.9.2-ubuntu64/bin写入到~/.bashrc文件末尾。 # 进入bin目录,查看当前位置 pwd 。根据当前路径,加在自己的$PATH:后面 source ~/.bashrc # 保存配置文件,使生效。 fastq-dump -h #
安装后很可能会报错: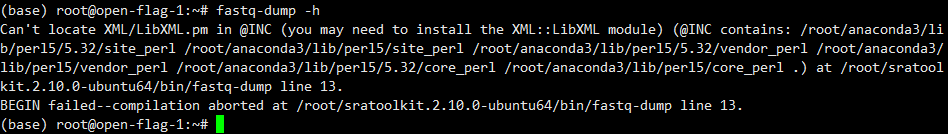
安装libxml-libxml-perl即可:
在单独创建的conda环境中安装就不会报错了。
https://grantm.github.io/perl-libxml-by-example/installation.html https://github.com/ncbi/sra-tools/issues/219 https://zoomadmin.com/HowToInstall/UbuntuPackage/libxml-libxml-perl
sudo apt-get update -y sudo apt-get install -y libxml-libxml-perl
conda安装sra-tools:
conda install sra-tools # sra-tools就是sratoolkits。
sratoolkits插件的作用有两个:下载SRA测序文件,将SRA测序文件转换为fastq格式。
安装生信软件时常见的报错原因:
SRA文件转fastq格式
由于下载的数据格式不符合分析所需格式,需转换。数据太多,写个脚本转换。这里是直接转换成立fastq.gz格式,很省内存,后面用这个格式文件分析就行。
如果文件数量少,一个个去转换。如果文件数量多,就需要批量转换。
mkdir scripts#创建一个文件夹用来存放脚本 mkdir fq#创建一个存放fq文件的文件夹 cd ../scripts cat > sra2fq.sh for i in ../sra.data/*sra do echo $i fastq-dump --split-3 --gzip ../sra.data/$i done cd ../fq nohup sh ../scripts/sra2fq.sh > ../logs/sra2fq.log 2>&1 & # nohub命令用于后台不断的运行命令不挂断,即使关闭终端也不停止运行。
SRA文件转fastq格式单个转:
# 单端测序 fastq-dump SRR14306907.sra -O ./ (结果生成:SRR14306907.fastq) fastq-dump --fasta SRR14306907.sra -O ./ (结果生成:SRR14306907.fasta) # 双端测序 fastq-dump SRR14306907.sra --split-3 -O ./ (结果生成:SRR14306907_1.fastq,SRR14306907_2.fastq) fastq-dump SRR14306907.sra --split-3 --gzip -O ./ (结果生成:SRR14306907_1.fastq.gz, SRR14306907_2.fastq.gz)
cutadapt
安装cutadapt:
mamba install cutadapt cutadapt -h #查看说明文档。
cutadapt使用:
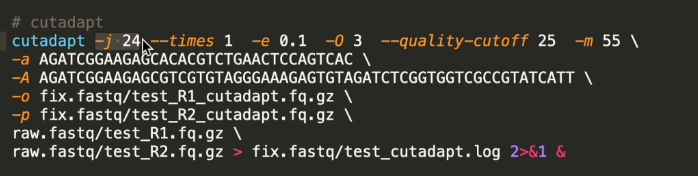
# cutadapt cutadapt -j 24 --times 1 -e 0.1 -o 3 --quality-cutoff 25 -m 55 \ # \是换行符。 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCA # adapter trimming reads1 -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT # adapter trimming reads2 -o ./test_R1_cutadapt.fq.gz \ # trim完的R1。 -p ./test_R2_cutadapt.fq.gz \ # trim完的R2。 ./test_R1.fq.gz\ # 需要trim的源文件。 ./test_R2.fq.gz > fix.fastq/test_cutadapt.log 2>&1 & #>是输出,这一步是输出log文件。2输出到1。2是标准错误,1是标准输出。 cutadapt参数 -j 24 # 调用CPU24个核。 --times 1 # 只进行一次去除。也就是我认为一条序列只会出现一侧adapter。 -e 0.1 # 当reads和adapter reads序列不一样的时候,我允许有10%的错误率,也就是10个碱基允许错配一个。 -o 3 #reads至少有3 bp和adapter匹配,我才认为是adapter。 --quality-cutoff 25 # 去除3端的低质量reads,一般选择25。 -m 55 # trim完以后,如果reads小于55,那么这一对reads就不要了。 3端的测序质量一定比5端的测序质量差。 reads2的测序质量一定比reads1的测序质量差。 reads如果要比对到参考基因组上,则reads在trim完以后不能小于40,不然比对的不准。 怎么知道adapter triming reads1/2是否正确呢? zcat test_R1.fq.gz | grep AGATCGGAAGAGCACACGTCTGAACTCCAGTCA #查看adapter序列是否在fastq序列里。按Esc退出。 linux下的top命令能够实时显示系统各个进程的资源占用情况,类似于windows系统的任务管理器。 cutadapt想要使用多核运算,一定要使用python3环境。 less -S test_cutadapt.temp.log # 查看log文件。
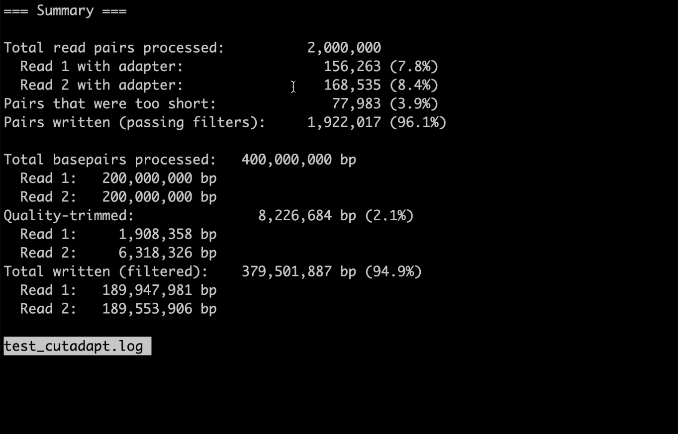
cutadapt运行完了以后的日志文件:
- read2 1 adapter占比7.8%
- reads2 adapter占比8.4%
- trim完以后,太短的占比3.9%
- 最后输出96.1%
illumina-adapter-sequences.pdf
当我们拿到一个fastQC报告时,首先看箱线图: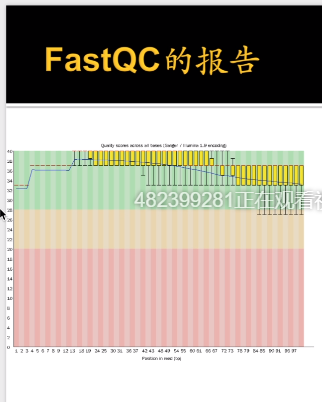
箱线图纵轴超过20,则表明测序质量还可以。超过30就算非常不错了。上图显示的测序质量就非常不错。
查看adapter比例: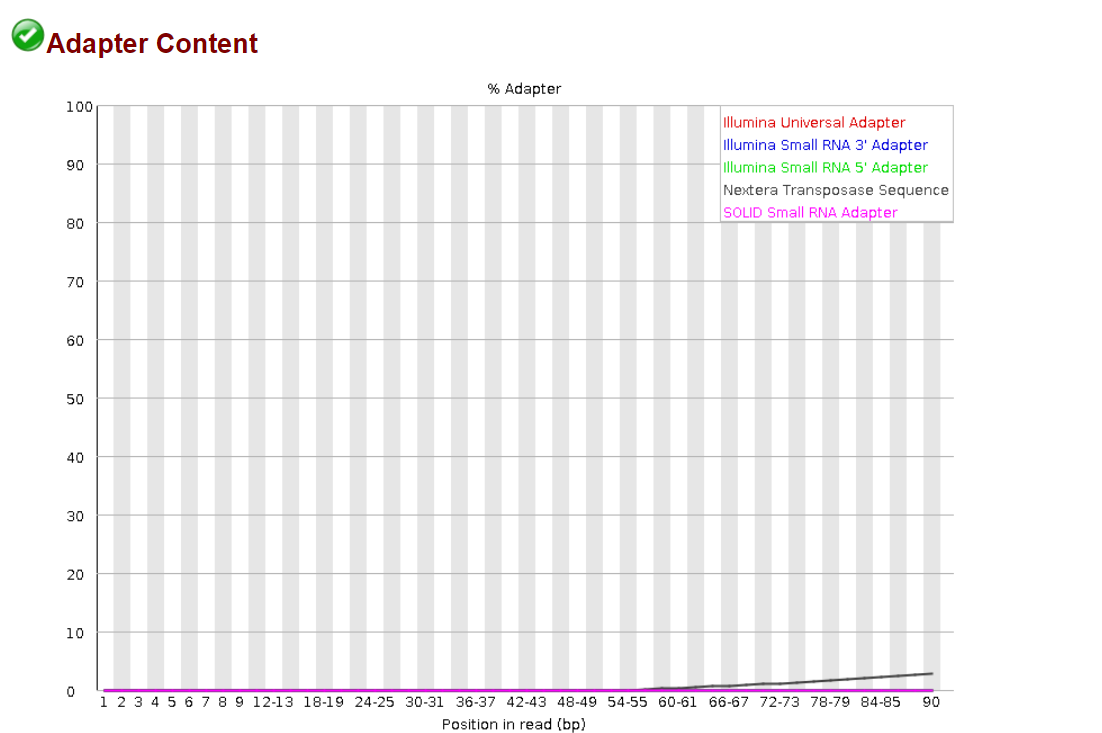
--quality-cutoff 25是啥?
碱基质量值怎么看?比如F,
fastq的标准有sanger、solexa、illumina1.3+、illumina1.5+等。目前最常见的是sanger标准。

万分之一的错误率,算出来Phred是40,千分之一是30,百分之一是20。
ASCⅡ表:0到255,每一个对应一个 字符。


phred值是不需要排序的,因为3端的测序质量是天然不如5端的。
参考资料
1、NASA 的 RNA-seq 标准流程代码
2、刘欣晔《分子生物学大实验》PPT:
2021细胞生物大实验刘昕晔正式上课课件.pptx